EN
中文
无论产品咨询、还是微流控相关技术探讨,欢迎与我们联系,我们有专业的技术支持为您提供服务。
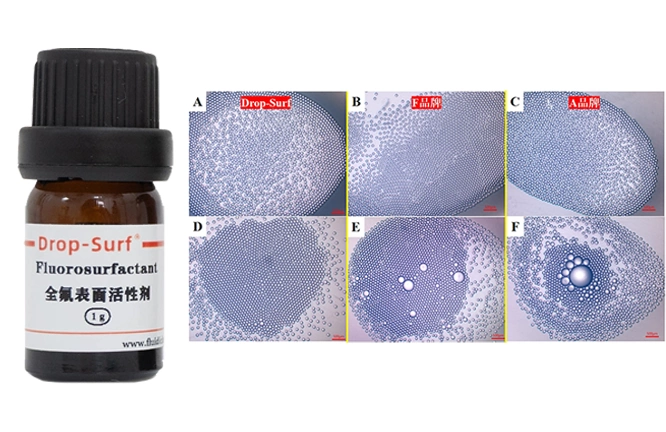
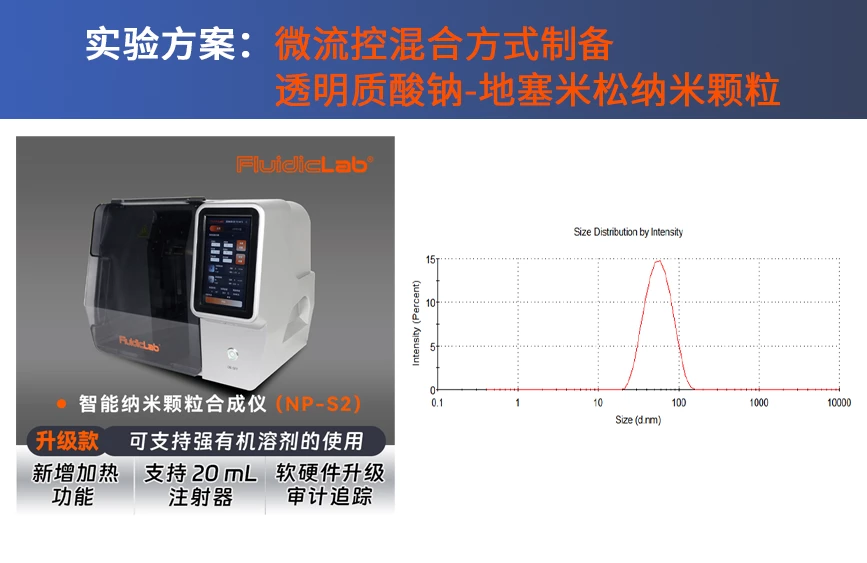
FluidicLab专注于研发生产用于液滴/微球和纳米颗粒应用的微流体组件和系统。我们的微流体系统可以用于许多不同的领域,如细胞生物学、化学、药物递送,化妆品和食品等,欢迎与我们合作,FluidicLab值得您信赖。
按需下载