目录(最新版本号V4.0)
- 实验方案:微流控混合方式制备包裹mRNA的脂质纳米颗粒
- 配制复方脂质-乙醇溶液
- 配制柠檬酸缓冲液
- 计算RNA浓度,配制 mRNA-柠檬酸缓冲液
- 微流控混合制备LNP(智能LNP合成仪S2的操作说明)
- 粒径与PDI的检测
- 产物的处理和保存
- 终产物的电位检测
- 实验方案:使用Qubit测定LNP包封率
- 实验方案:使用酶标仪测定LNP包封率
- 实验方案:LNP转染293T细胞
- 附录
| 温馨提示:由于实验方案在不断优化更新,以下实验方案内容仅供参考,如有需要,请通过网站上的联系方式向工作人员获取实时更新的版本PDF文件。(FluidicLab编辑并发布,转载请通知我们)。 |
实验方案:微流控混合方式制备包裹mRNA的脂质纳米颗粒(mRNA-LNP的制备)(当前为最新V4.0版本)
实验目的:
参照 Moderna、BioNtech 和 Alnylam 的新冠疫苗配方,以SM102、ALC-0315、MC3为主要阳离子脂质制备包载 mRNA 的脂质纳米颗粒(lipid nanoparticles, LNPs)。
实验原理:
ALC-0315、 MC3、和 SM102 是三种可用于人体的脂质。在酸性条件下,质子化形成阳离子脂质,能够通过静电作用和带负电的 mRNA 结合。脂质与溶有mRNA的水性溶液混合后析出,自组装形成载有 mRNA 的脂质纳米颗粒。本实验中采用微流控混合法,让脂质溶液与mRNA溶液在微混合器中充分、迅速、高度可重复地形成粒径均一可控的LNP。在制备过程中,脂质溶解于乙醇,核酸溶解于酸性缓冲液中,故制备得到的LNP初产物含有高浓度乙醇。因此后续还需要透析或者超滤去除残余的乙醇并将溶液体系置换至中性缓冲液中,以备后续生物学实验及长期保存。
实验材料:
| 试剂 | 复方磷脂成分 (溶于无水乙醇) | ALC-0315 、DLin-MC3 、SM-102 |
| DSPC | ||
| DMG-PEG2000/ALC-0159 | ||
| CHOLESTEROL | ||
| 缓冲液成分 (溶于超纯水) | 柠檬酸(C6H8O7•H2O) | |
| 柠檬酸钠(C6H5Na3O7•2H2O) | ||
| 溶剂 | 无水乙醇 | |
| 超纯水 | ||
| 置换所需缓冲液 | 1 x PBS 缓冲液(pH 7.4)或 20 mM Tris 缓冲液(pH 7.4) | |
| 耗材 | 合成需要 | BD/AD/KDL 带鲁尔口的注射器若干[1] |
| Falcon 15 ml离心管(352096) | ||
| 若透析 | Thermo Scientific™ Slide-A-Lyzer 透析盒 (20K MWCO)[66003] | |
| 若超滤 | Millipore 15ml/30 kd 超滤离心管(外径50ml尺寸)[UFC903096] | |
| 微混合芯片 | 微混合芯片 | FluidicLab LNP-B1 |
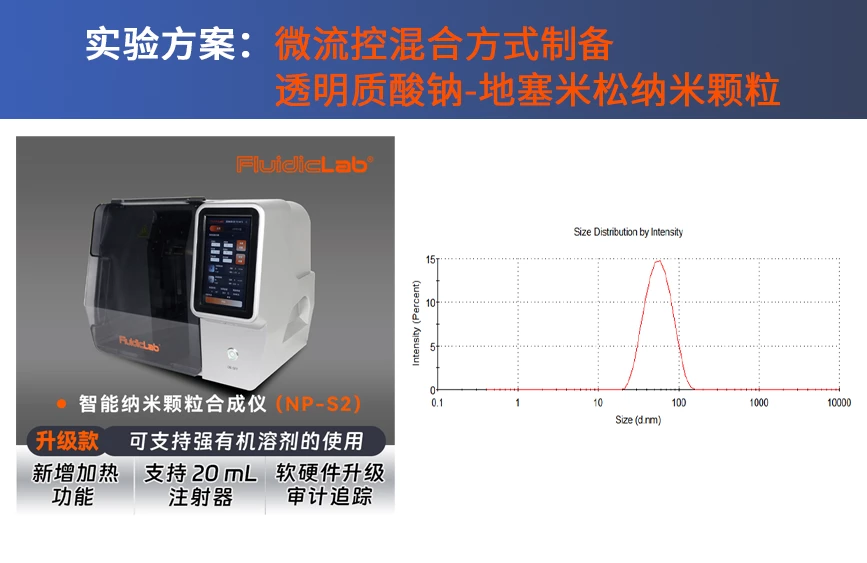
| 设备 | 微流控LNP合成 | FluidicLab 智能纳米颗粒合成仪(型号:NP-S2) |
| LNP检测 | 动态光散射仪(本文中使用的为 Malvern ZS90) | |
| LNP超滤 | 冷冻离心机 | |
| 核酸包封检测 | Qubit 4.0 或 酶标仪等检测仪器[2] |
[1]. 经测试,该三款注射器推杆较硬,推送液体较为准确。1mL AD/KDL注射器无需鲁尔口;
[2]. 此处不推荐NanoDrop检测核酸浓度,其对降解核酸以及低浓度核酸检测时会有较大波动。
实验步骤
1. 配制复方脂质-乙醇溶液:LNP的成分与分子量见下表:
(附:LNP合成质量检测、转染细胞等实验方案)插图")
所有成分总浓度配制成 12 mM(约7.5 mg/ml)。如果需要摸索脂质浓度对 LNP 最终粒径的影响条件,可以配制以下浓度:8 mM(约 5 mg/ml)、12 mM(约 7.5 mg/ml)和 16 mM(约 10 mg/ml)等进行测试。需注意,低于4mM会难以形成均一的LNP,而过高浓度的磷脂反而会导致LNP粒径的增大。不同脂质浓度对LNP粒径的影响见 附录-附件1。我们推荐使用12mM浓度的复方脂质作为实验的起始条件。
备注 :配制好的磷脂溶液可于4℃密封保存半年。再次使用前请平衡至室温,直至溶液澄清。阳离子磷脂保存条件比较苛刻 ,常温下接触空气后会发生氧化变质,受潮则易析出。直接结果为合成 LNP时粒径出现异常增大(如粒径增大一倍以上),请确保妥善密封保存。
您也可以选择购买FluidicLab的LNP(脂质纳米颗粒)包封试剂盒。出厂前我们进行了严格的LNP合成检测,以确保LNP合成的质量和稳定。
2. 配制柠檬酸缓冲液
使用超纯水分别配制 100 mM 的一水合柠檬酸(分子量:210.14 ,称取 1.05 g)和二水合柠檬酸钠(分子量:294.10 ,称取 1.47 g)溶液各 50 ml 。取 33.0 ml 柠檬酸溶液和 17.0 ml 柠檬酸钠溶液混合,用NaOH调至pH =4 。随后用超纯水定容至 100 ml,加入终浓度0.1% 的DEPC静置30分钟。 高压灭菌去除 DEPC,即得 50 mM柠檬酸缓冲液。柠檬酸缓冲液与乙醇配制的磷脂,需要分别用0.22 μm的MCE滤膜(柠檬酸缓冲液)与PTFE滤膜(乙醇配制的磷脂)分别过滤,确保其终产物不含微小的固态颗粒(包封试剂盒内含100mM柠檬酸缓冲液,稀释后可直接使用)。
3. 计算RNA浓度,配制 mRNA-柠檬酸缓冲液:
氮磷比(N/P)是mRNA-LNP包封中常用的计算核苷酸用量与磷脂用量的参数。每个碱基含有一个磷酸根,1 mol的RNA或DNA即含有1mol的磷酸根(P)。磷脂中仅以可电离脂质比例计算氮原子数,每摩尔复方脂质中含有 0.5 mol氮原子(N)。
备注:虽然大多数的配方推荐的N/P比为6,但是实验表明,在后续的混合和超滤过程中,会损失一定的磷脂。所以在实践中,我们推荐您(可根据实验需要)将N/P比提高为8。更高的N/P比可以降低超滤和透析引起的粒径变大,并提高 mRNA 的利用率。缺点是可能会增大对细胞的毒性 ,降低细胞的存活率。对于包封率,高N/P比也并无优势。
核糖核苷酸的平均分子量为339.5,形成RNA脱水聚合后NEB推荐计算均值为320.5 g/mol。脱氧核糖核苷酸的平均分子量为327.0,形成双链DNA脱水聚合后,NEB推荐计算均值为308.0 g/mol。以可电离脂质比例计算氮原子数,每摩尔复方脂质中含有 0.5 mol氮原子。
(附:LNP合成质量检测、转染细胞等实验方案)插图1")
以柠檬酸缓冲液/复方磷脂流速比(FRR)= 3、N/P = 6、磷脂浓度 = 12 mM为例,进行核酸使用量的计算:
柠檬酸钠缓冲液中RNA浓度:(26.71 × 12) / 3 = 106.84 ng/μL;
柠檬酸钠缓冲液中DNA浓度:(25.67 × 12) / 3 = 102.68 ng/μL。
常用复方磷脂浓度与缓冲液中核酸浓度对应表
| 脂质浓度(mM) | 8 | 12 | 16 | 8 | 12 | 16 |
| N/P | 6 | 8 | ||||
| FRR | 3 | 3 | ||||
| ssRNA 浓度(ng/μl) | 71.23 | 106.84 | 142.45 | 53.41 | 80.12 | 106.83 |
| dsDNA 浓度(ng/μl) | 68.45 | 102.68 | 136.91 | 51.33 | 77 | 102.67 |
4. 微流控混合LNPx(智能LNP合成仪S2的操作说明):
(附:LNP合成质量检测、转染细胞等实验方案)插图2")
FluidicLab智能纳米颗粒合成仪(NP-S2)
在制备纳米颗粒(如LNP、Liposome)的过程中,关键步骤是有机相与缓冲液在微流控芯片中高速均匀的混合。通过先进的微流控技术,智能纳米颗粒合成仪(NP-S2)能够制备粒径高度均一且可控的纳米颗粒。经验证,S2同时具备了高重复性、低样本消耗、易于操作等优势,大幅提高了客户前期配方筛选的效率。
4.1. 装配芯片。
将微流控芯片(B1-含鲁尔口)的鲁尔口朝上,装入适配器中(图1. A~C)。
(附:LNP合成质量检测、转染细胞等实验方案)插图3")
图1. A.芯片适配器和微流控芯片;B. 微流控芯片鲁尔口朝上装入适配器中;C. 装配完成;
4.2. 装配反应仓
将B1芯片鲁尔口朝内朝下,适配器把手朝外,装入微流控反应仓(图2. A~B)
(附:LNP合成质量检测、转染细胞等实验方案)插图4")
图2. A~B: B1芯片与适配器装入反应仓的操作。
4.3. 预冲洗芯片
实验前的预冲洗步骤是十分关键且必要的,请务必将芯片中预先填充乙醇和缓冲液,以帮助LNP高质量地合成。
4.3.1.注射器套筒的装配(此处以5ml注射器套筒为例)。
握住芯片适配器把手,向上转动反应仓(图3.A); 转动后可方便注射器适配器(套筒)的装入(图3. B)。随后装入符合需求的注射器套筒(使用20 ml注射器无需额外套筒)(图3. C)。
(附:LNP合成质量检测、转染细胞等实验方案)插图5")
图3. A. 反应仓握持及转动方向示意图; B. 转动完成示意图;C. 装入符合需求的注射器套筒(使用20 ml注射器无需额外套筒)。
4.3.2. 注射器及收集管的装配(此处以5ml注射器为例)。
用两支注射器吸取乙醇与缓冲液,并排空气泡(图片未展示)。注意注射器吸取液体必需略大于实际实验用量。以拇指与食指握住注射器,挂耳对准自己(图4. A),向上插入反应仓套筒。向上插入稍稍遇到阻力后,在向上用力的同时旋转注射器(图4. B)。注意此处旋转并非是为了螺纹旋紧,而是释放对接中的应力,使得注射器与芯片连接更加牢固。装配完注射器后,将反应仓向下转动归位(图4. C),此时注射器与水平面垂直。最后安装两个15 ml离心管作为产物、废液收集管(图4. D)。安装部分操作完毕。
(附:LNP合成质量检测、转染细胞等实验方案)插图6")
图4. A. 注射器装配时有利于推入的握持方式; B. 注射器装配时的用力方向示意图;C. 归位后反应仓与注射器形态示意图; D. 安装完成的收集管与废液管。
4.3.3. 软件的设置
首先连接设备电源。注意设备背后电源线处有一总开关。打开总电源开关后,打开前方控制面板电源。随后进行注射器参数及运行程序的设置。点击屏幕右上角注射器设置。依次选择脂相和水相注射器参数(图5.A)。正确的注射器参数才能保证准确的液体推送。请务必每一次实验前检查注射器参数是否与本次实验所用注射器一致。随后返回主页面。
(附:LNP合成质量检测、转染细胞等实验方案)插图7")
图5. A. 注射器参数的设置。
在主界面(图5最右)依次设置流速比,总流速,产物总体积,前后废液等参数。预冲洗推荐参数:流速比
1:1;总流速16 mL/min;产物总体积3 mL。预冲洗可不进行产物收集,产物体积设为0 mL即可。前废液设置为3mL设置完成点击界面最下端“开始”图标,即可开始预冲洗。
4.3.4. 微流控混合制备LNP
1) 装配或更换适合的注射器套筒;
2) 使用注射器吸取配制好的复方磷脂及mRNA-柠檬酸缓冲液,排空气泡并与芯片连接。推荐最低吸取0.5ml;且吸取液体体积比实验计算用量略大0.1~0.2ml。如以1:3的流速比计划合成1ml终产物,0.2ml前废液( 即总产物1.2 ml )。则至少吸取0.5 ml( >0.3 ml,至少0.5ml )的复方磷脂和1 ml( > 0.9 ml )的mRNA-柠檬酸缓冲液。
3) 装配注射器,并于软件中修改或确认注射器品牌及参数。
4) 设置合成参数。我们推荐初始合成参数为:流速比1:3; 总流速12 ml/min,前废液0.2 ml,后废液0 ml。对于B1芯片:一定区间内流速越大,粒径越小,PDI也会越稳定。具体流速-粒径/PDI对应关系,参数详见附录-附件2实验数据。
5) 点击“开始”图标进行实验,NP-S2智能合成仪即可全自动切换前(后)废液并收集产物。
5. 粒径和PDI的检测
mRNA-LNP产物可以使用动态光散射仪测定 LNP 粒径和均一度。正常结果如下图所示:
(附:LNP合成质量检测、转染细胞等实验方案)插图8")
值得注意的是,由于LNP初产物中含25%乙醇,高浓度乙醇会导致直接检测初产物粒径有80%以上的偏差。因此需要及时地使用柠檬酸缓冲液将初产物稀释5倍进行检测。我们的实验数据显示,将乙醇的浓度稀释至5%以下,所测量的粒径才相对较为准确(误差<10%)。具体的稀释数据参考 附录-附件3 《乙醇浓度对LNP粒径及PDI检测的影响》。
备注:LNP初产物中高浓度乙醇会造成LNP融合,故推荐合成后立即用缓冲液稀释。若进行透析则无需稀释,尽快(10min内)将产物置于透析袋和透析液中进行透析。
若遇终产物粒径/PDI异常,合成后的立即检测数据有助于排查原因。实验论文中对外展示的粒径/PDI数据一般是经过后处理的终产物数据。
6. 产物的处理和保存
可以根据实际情况选择超滤或者透析来去除产物中的乙醇并置换缓冲液。如果希望获得LNP粒径更小的终产物,则推荐进行透析。具体对比见 附录-附件4。
6.1.透析
1. 产物在制备后用注射器直接将产物移入透析卡(推荐使用Thermo的Slide-A-Lyzer™ 透析盒,20 K MWCO)或20 KD预处理过的透析袋。
2. 将装有LNP初产物的透析卡或透析袋置于至少50倍体积1 x PBS(pH=7.4),或50倍体积20 mM Tris-HCl(pH=7.4)于4℃透析2h。
3. 将透析液更换为50倍体积,含有10% 蔗糖 的PBS(pH=7.4),或50倍体积,含有8% 蔗糖的20 mM Tris-HCl(pH=7.4)于4℃透析过夜。
4. 透析后LNP终产物可于4℃或-20℃进行保存。
6.2. 超滤
1. 样品收集后 ,加入3倍体积的柠檬酸缓冲液稀释。
备注 :不建议直接使用 PBS 或者 Tris 缓冲液稀释 ,过快改变 LNP 外环境 pH 会造成不可控的粒径/PDI的增大。
2. 使用 Milipore 100 kD 超滤管,3000 g离心10 min,超滤至原体积的 1/4。(使用过小孔径的超滤管会使得超滤困难,超滤时间延长。不建议使用10kD或更小的超滤管。)
3. 补加1 x PBS(或20 mM Tris-HCl)至原体积 ,再次超滤10 min,至原体积 1/4。
4. 重复步骤 3 两次 ,将乙醇含量降低至 0.5%以下。
5. 最后一次使用含有蔗糖保护液的缓冲液超滤浓缩,使得PBS缓冲液的终产物中含有10%的蔗糖(Tris-HCl缓冲液终产物含有8%蔗糖)。
6. 将超滤后的液体收集,于4℃ 或 -20℃冻存。
Tips :
LNP保存缓冲液与粒径的关联:
虽然根据 Moderna 公开数据显示 ,SM-102 适合使用含有8%蔗糖 的 20 mM Tris buffer(pH=7.4) 进行保存,但是我们测试发现DLin-MC3-DMA,SM102两种配方使用含有10% 蔗糖的PBS(pH=7.4)透析后粒径更小,且差异显著。ALC-0315配方生成的LNP保存于两种缓冲液中,粒径/PDI接近。故,对粒径有要求的实验者建议使用含有蔗糖的PBS进行透析和保存。
LNP保存时效与保存环境的关联:
LNP-RNA产物于含有10 % 蔗糖的PBS中,-20℃ 条件下保存一个月,与新制备LNP-RNA产物比较,稳定性和体内效力两者接近[1]。我们的实验结果则显示,4℃保存一个月,LNP产物粒径/PDI基本稳定,且仍具有转染细胞的能力。故,请4℃或-20℃保存。切勿进行-80℃冻存[1]。
LNP转染效率与保存缓冲液的关联:
有文献报道显示DLin-MC3-DMA于TBS(Tris-buffered saline)缓冲液条件下转染效率会高于PBS缓冲液[2]。所以若对后续转染细胞或小鼠有需求的实验者,可以参考该文献对缓冲液体系进行优化。
7. 终产物的电位检测
得到终产物后,除了检测粒径、PDI等常规参数外,也可对产物进行Zeta Potential的检测。
1.100 μL终产物加900 μL水或PBS定容至1mL,混匀后加入马尔文DTS1070电位比色皿进行检测。正常检测结果如图所示
使用PBS测量时,由于液体中的电导率过高,会导致测量结果无峰图显示。马尔文粒径仪并不适合高盐条件下的检测。但根据马尔文工程师的反馈,该现象并不会影响电位测量的准确性。若要获得相应的电位分布图,则建议使用ddH2O进行稀释。若仍需回收利用,则建议使用PBS进行稀释。我们的测试结果显示,使用PBS对终产物进行不同倍数的稀释后,产物Zeta Potential无显著变化(附件5)。
2.若需要重复利用内部液体,则使用1mL注射器将内部液体吸出。加入液体检测时,需确保加入的液体没过电极片。详细电位测量细节,可参考Malvern ZS90说明书。常规LNP配方的电位检测峰图如下:
(附:LNP合成质量检测、转染细胞等实验方案)插图9")
[1]. Kim B, Hosn RR, Remba T, Yun D, Li N, Abraham W, Melo MB, Cortes M, Li B, Zhang Y, Dong Y, Irvine DJ. Optimization of storage conditions for lipid nanoparticle-formulated self-replicating RNA vaccines. J Control Release. 2023 Jan;353:241-253.
[2]. Henderson MI, Eygeris Y, Jozic A, Herrera M, Sahay G. Leveraging Biological Buffers for Efficient Messenger RNA Delivery via Lipid Nanoparticles. Mol Pharm. 2022 Nov 7;19(11):4275-4285.
实验方案:使用Qubit测定LNP包封率及利用率
实验目的
对制备的 LNP 包封 mRNA 的效果进行测定。
实验试剂
| 试剂名称 | 货号 | 品牌 |
| Qubit™ RNA HS Assay Kit | Q32852 | Invitrogen |
| Triton-X-100 AR级 | T824275-500ml | 麦克林 |
| DEPC水或TE buffer | / | / |
实验耗材
| 名称 | 货号 | 品牌 |
| Axygen 0.5ml离心管 | MCT-500-C | Axygen |
| 枪头若干 | / | / |
实验仪器
Qubit 4.0 Invitrogen
实验步骤
检测步骤可参考Qubit™ RNA HS Assay Kit 说明书:Document Connect (thermofisher.cn)
操作步骤简述如下:
1)配制RNA检测工作液:吸取 [(样品数)+( 2 标曲)+( 1 )] μl的Qubit™ RNA HS reagent,加入200 ×([(样品数)+( 2标曲)+( 1 )] μl的Qubit™ RNA HS buffer,配成1:200比例配制工作液。
【如6个待测样本,吸取(6+2+1)= 9 μl的Qubit™ RNA HS reagent,加入200 x (6+2+1) μl = 1800 μl的Qubit™ RNA HS buffer中配成工作液。】
2)制作常规标曲,并于Qubit 4.0上进行标曲的校准。
3)检测LNP合成终产物中,游离RNA含量。
【若浓度过低无法检测,则吸取190μl工作液,加入10μl待测样本,充分混匀后检测。在Qubit 4.0中选择1μl选项,并随后将数值÷10,进行10倍换算。】
4)使用TE buffer或DEPC水配制4 % Triton X-100,与LNP终产物1:1混合(5μl+5μl即可)。使用该终浓度为 2 %的Triton X-100破乳5分钟。
5)在常规标曲中添加 1 μl的 2 % Triton X-100,并于Qubit 4.0上进行含有Triton X-100新标曲的校准[3]。
6)测量LNP破乳后的RNA浓度。
【注意该RNA溶液在破乳时稀释了1倍。真实浓度需要 × 2。】
7)进行包封率的计算,公式为:
载药量 = 破乳后读值 - 破乳前读值
包封率(%) = [载药量/破乳后读值] × 100%
8)同时可进行RNA利用率的计算,公式为:
RNA利用率 (%) = [载药量/RNA投入量] × 100 %
【3】经检测,Triton X-100的存在会影响RNA检测的准确度, 故推荐制作新标曲。
实验方案:使用酶标仪测定LNP包封率
实验目的
对制备的 LNP 包封 mRNA 的效果进行测定。
实验原理
Quant-iT™ RiboGreen® RNA 试剂是一种超灵敏的荧光核酸染色剂 ,可检测溶液中 1-200 ng 的核酸 ,这种核酸染料无法透过 LNP ,因此只有游离的未被 LNP 包载的核酸可 以被结合。Triton-100 作为一种表面活性剂常被用做破乳剂,使用 1%的 Triton-100 处理 获得的 LNP-mRNA 可以使包载的核酸释放 ,得到总核酸量。通过计算破乳前后核酸量的
差异得到载药量 ,再除以总核酸量即可得到包封率 ,即:
包封率(%) =(破乳后定量-破乳前定量)/破乳后定量
实验材料
Quant-iT™ RiboGreen™ RNA 检测试剂盒(Thermo Fisher , R11490)
Triton™ X-100 ( SIGMA-ALDRICH ,T8787-100ML)
DEPC 水
CellCarrier-96 Ultra Microplates ( PerkinElmer , 6055308)
实验步骤
试剂准备等检测步骤细节可参考Quant-iT™ RiboGreen® RNA 试剂盒说明书。
1) 将 20xTE 用 DEPC 水稀释至 1x;用稀释好的 1XTE 稀释试剂盒中的标准品,稀释成 2 μg/mL 及 100 ng/mL 两种终浓度;
2) 用 1xTE 稀释试剂盒中的 QuantiT™ RiboGreen® Reagen,稀释成 200 倍及 2000倍两种终浓度;注意要同时制作含有/不含有Triton X-100的两套标曲;
按照下表在 CellCarrier-96 Ultra Microplates 中加入以下试剂 ,做复孔:
High range
(附:LNP合成质量检测、转染细胞等实验方案)插图10")
Low range
(附:LNP合成质量检测、转染细胞等实验方案)插图11")
3) LNP 破乳。将 Triton-100 用 1xTE 稀释到 2 %,取 100 μl 加入 CellCarrier-96 Ultra Microplates, 加入 1 μl 制备好的 LNP,处理 5 分钟,加入 100 μl 200-fold diluted quant-iT RiboGreen Reagent。
4) 4. LNP 游离核酸测定。在 CellCarrier-96 Ultra Microplates 中加入 100 ul 1xTE ,取 1 μl 制备好的 LNP 加进去 ,混匀 ,加入 100 μll2000-fold diluted quant-iT RiboGreen Reagent。
5)读板使用 SPARK 读取荧光强度 ,激发光设置为 480 nm ,发射光为 520 nm6. 数据处理
6) 标曲制备
(附:LNP合成质量检测、转染细胞等实验方案)插图12")
7) 样品定量
载药量 =破乳后读值-破乳前读值
包封率(%) =载药量/破乳后读值
8)同时可进行RNA利用率的计算,公式为:
RNA利用率 (%) = [载药量/RNA投入量] × 100%
实验方案:mRNA-LNP转染293T细胞
实验目的
以 293T 细胞为例 ,使用包封了 EGFP-mRNA 的 LNP 对细胞进行转染。
实验步骤
1.HEK-293T 细胞消化。
选取 P3 – P15之间的HEK-293T细胞[4]。弃上清后PBS清洗。1 ml 胰酶37℃消化1min。使用1 ml 含10 % FBS的DMEM终止消化。轻柔吹打细胞,收集细胞悬液。
2.细胞铺板
室温125 x g,离心5~10 min。弃上清,并以3ml,noPS,10 % FBS的DMEM重悬细胞。对细胞进行计数。取5 x 104 个细胞/24孔板单孔铺板。每个孔中添加 1 ml noPS , 含10 % FBS的DMEM培养基。
3.细胞转染
可用lipo作为阳性对照进行转染实验。对于LNP转染,直接加入包封完成的EGFP-mRNA-LNP即可。每孔添加500ng。若使用其他规格培养皿或培养板,保证包封在LNP内的EGFP-mRNA-转染量达到终浓度500ng/ml即可。培养24h后即可观察到荧光。
若mRNA-LNP浓度较高,可在转染前使用opti-MEM对LNP进行稀释或定容。
4.检测
在 转染48 小时后 ,收集细胞 ,流式检测 GFP 信号阳性率。
细胞转染结果见 附录-附件5
[4].大量的实践结果表明,293T细胞被支原体感染后,虽不影响Lipo-mRNA的转染,却会极大降低mRNA-LNP的转染效率。这会导致阳性对照正常,转染实验组无表达。请在转染前,务必进行细胞支原体检测,确保细胞的状态。
附录
附件清单
- 附件1:复方磷脂浓度与粒径/PDI的关系图;
- 附件2:LNP合成总流速与LNP粒径/PDI的关系图;
- 附件3:乙醇浓度对LNP粒径及PDI检测的影响;
- 附件4:LNP合成超滤与不同条件透析对比
- 附件5:LNP不同稀释倍数对Zeta电位的影响
- 附件6:mRNA-LNP转染HEK-293T
附件1:复方磷脂浓度与粒径/PDI的关系图
(附:LNP合成质量检测、转染细胞等实验方案)插图13")
实验条件: 仪器:Fluidiclab-S1 ;芯片:Fluidiclab-B1
MC3复方脂质配方;总流速:12 ml/min;流速比(脂相:水相)= 1 : 3;前废液0.2 ml;LNP空包无mRNA;初产物未经过柠檬酸缓冲液稀释。可见12 mM合成粒径最小。
附件2:LNP合成总流速与LNP粒径/PDI的关系图
(附:LNP合成质量检测、转染细胞等实验方案)插图14")
实验条件:仪器:Fluidiclab-S1 ;芯片:Fluidiclab-B1
MC3配方:12 mM浓度;流速比(脂相:水相)= 1 : 3;前废液 0.2 ml;LNP空包无mRNA;初产物经柠檬酸缓冲液稀释5倍后检测;可见速度越高合成LNP粒径越小。
附件3:乙醇浓度对LNP粒径及PDI检测的影响
LNP越稀释粒径越小?
LNP全称Lipid Nanopartical(纳米脂质颗粒)。常用LNP合成仪,利用微流控的方式在微流控芯片中进行高度可控的合成。LNP合成后,有一些老师发现使用马尔文粒度仪测量LNP粒径时,对LNP产物做了稀释的粒径小于LNP产物直接进行检测。这是怎么回事呢?
众所周知,马尔文粒度检测仪对于粒子粒径大小的检测基于粒子的布朗运动速率。所以液体的折射率/粘度等参数都会对检测结果有所影响。这也是在进行粒度检测之时需要选择粒子所在溶液及液相参数的原因。
那不禁想问,LNP合成后所残存的乙醇是否对LNP粒径和PDI有影响?产物直接做检测是否得到的是真实的粒径?为了回答这些问题,我们进行了一系列验证实验。
25%乙醇会显著增加粒径的检测值
一般LNP合成时,有机相(溶解了脂质的乙醇):水相(溶解了mRNA的柠檬酸钠缓冲液)的流速比为1:3。这就使得产物会有25%的乙醇。为了模拟这个乙醇浓度,我们在标准品70nm的单分散交联聚苯乙烯标准微球(即70nm PS微球)中加入了25%乙醇进行检测。与原始溶液中的标准微球相比,添加终浓度25%乙醇后,检测到的粒径值接近130nm,几乎是原始粒径的一倍(图1.A);而相比之下,添加终浓度为25%的柠檬酸钠缓冲液中微球粒径和PDI与原始检测结果并无差异(图1.A)。
(附:LNP合成质量检测、转染细胞等实验方案)插图15")
(附:LNP合成质量检测、转染细胞等实验方案)插图16")
图1:标准微球的乙醇稀释实验。A:终浓度为25%的柠檬酸缓冲液和25%的乙醇对70nm标准微球的稀释测量结果;B:标准微球的二度稀释实验。Error bar为样本标准差;N.S:无显著差异;检验方式:Student’s t-test。
乙醇改变粒径检测值而不改变其实际大小
熟悉材料学的朋友可能会提出异议,PS是会被乙醇降解的。这个结果是否是由于乙醇将PS微球溶解从而导致检测粒径增大呢?为了回答这个问题,我们将上一步实验得到的包含PS微球的25%的乙醇溶液再次进行了稀释,将乙醇浓度降低至5%。结果发现,检测粒径重新变小,,且与直接加入5%的乙醇相比,粒径数值没有显著差异(图1.B)。粒径可能被溶解从而增大,但并不会自发缩小。该结果可以认为,粒径数值上的改变完全是因为乙醇浓度造成的影响。
推荐LNP合成后进行5倍稀释(检测时,乙醇终浓度≤5%)
意识到乙醇的存在会对粒径检测产生剧烈影响,那究竟在LNP合成后,稀释多少倍进行检测比较合适呢?我们在标准微球中加入了1%~25%的乙醇进行了反复测试,观察乙醇对粒径测量的影响。结果发现,即使2%的乙醇即可使得粒径测量值有着显著的增加(图2)。并且随着乙醇浓度的逐渐增加,标准微球的测量数值逐渐增加。但是根据图1.B的结果我们可以知道,这仅仅是检测数值的变化,并非粒子真实的粒径改变。大家可以根据实际需要,参考我们的结果,进行LNP合成溶液的稀释和粒径检测。
(附:LNP合成质量检测、转染细胞等实验方案)插图17")
图2:70nm PS微球的梯度稀释实验。Error bar为样本标准差;N.S:无显著差异;检验方式:Student’s t-test。
虽然乙醇浓度越低越接近真实的粒径值,但是我们仍推荐将乙醇浓度降低至5%附近进行检测。不做进一步稀释的原因是,过高的稀释倍数会导致溶液环境的剧烈变化。尤其是高倍的PBS稀释,会剧烈改变溶液的pH值。这将直接导致粒子电位的剧烈变化,从而使得LNP变得不稳定,PDI异常增大(图3)。过大的稀释倍数也会同时使得LNP粒子浓度的下降,这将会大大增加马尔文粒度检测仪的检测时长,降低精准度。5倍左右的稀释既可以保证与真实粒径的差异在15%以内,且PDI不会有剧烈波动。
(附:LNP合成质量检测、转染细胞等实验方案)插图18")
图3:LNP稀释倍数与粒径/PDI的关系。
LNP合成条件:脂相: Alnylam配方(Dlin-MC3-DMA/DSPC/cholesterol/PEG-2000-DMG=50/10/38.5/1.5,mol/mol),12mM脂质总浓度;水相:柠檬酸缓冲液(50mM柠檬酸+50mM柠檬酸钠,pH=4.0),无mRNA;脂:水FRR(流速比)=1:3;总流速=12ml/min。使用仪器:FluidicLab(上海澎赞生物)LNP智能合成仪-S1。芯片:FluidicLab(上海澎赞生物)B1(鱼骨)芯片。
附件4:LNP合成超滤与不同条件透析对比
(附:LNP合成质量检测、转染细胞等实验方案)插图19")
实验条件:仪器:Fluidiclab-S1 ;芯片:Fluidiclab-B1
MC3配方:12 mM浓度;流速比(脂相:水相)= 1 : 3;前废液 0.2 ml; LNP空包无mRNA;
合成初始——初产物经柠檬酸缓冲液稀释5倍后检测;
超滤使用Millipore 15ml/30 kd 超滤离心管(外径50ml尺寸)[UFC903096]。
透析使用Thermo Scientific™ Slide-A-Lyzer 透析盒 (20K MWCO)[66003]。
超滤后浓缩则使用[66003]透析盒后再使用[UFC903096]进行浓缩。实验步骤参照第一节:《实验方案:微流控混合方式制备包裹mRNA的脂质纳米颗粒(mRNA-LNP的制备)》。
附件5:LNP不同稀释倍数对Zeta电位的影响
(附:LNP合成质量检测、转染细胞等实验方案)插图20")
实验条件:仪器:Fluidiclab-S2 ;芯片:Fluidiclab-B1
MC3配方:12 mM浓度;流速比(脂相:水相)= 1 : 3;前废液 0.3 ml;
柠檬酸缓冲液中mRNA浓度106.84 ng/μL或不含mRNA(unloaded)
产物使用PBS稀释定容至1mL后检测
zeta电位检测参数设置:材料吸光度0.001;材料折射率 (RI)1.45;分散剂折射率(RI)1.33 ;粘度0.8872 cP。
附件6:mRNA-LNP转染HEK-293T
(附:LNP合成质量检测、转染细胞等实验方案)插图21")
(附:LNP合成质量检测、转染细胞等实验方案)插图22")
(附:LNP合成质量检测、转染细胞等实验方案)插图23")
实验条件:见:第5节:《实验方案:mRNA -LNP转染293T细胞》。